Općenitost
Uvjet pigmentozni retinitis (RP) identificira skupinu genetskih bolesti koje karakterizira progresivna degeneracija retine.

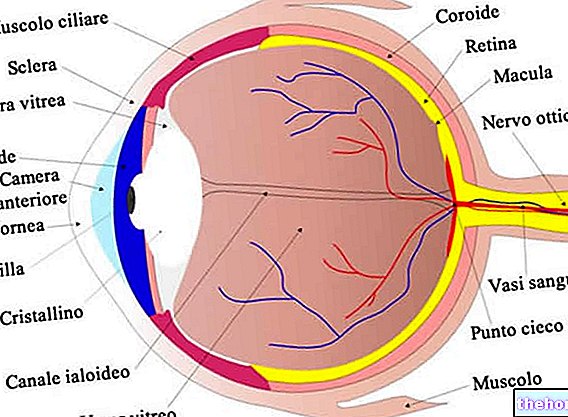
Retinitis pigmentosa retinalna je distrofija karakterizirana postupnim gubitkom fotoreceptora i disfunkcijom pigmentnog epitela, što znači da retina progresivno smanjuje svoju sposobnost prijenosa vizualnih informacija u mozak putem vidnog živca.
Patološki proces započinje promjenama pigmentnog epitela mrežnice. Kako pigmentozni retinitis napreduje, dolazi do stanjivanja krvnih žila koje opskrbljuju mrežnicu, a koje podliježu atrofiji. Nakon pregleda fundusa, karakteristične naslage se vizualno detektiraju. Retinalni pigment (retinalni pigment) otuda i naziv po bolesti). Atrofične promjene i oštećenja također mogu zahvatiti vidni živac i postupno odumiru fotoosjetljive stanice mrežnice.
Pacijenti pogođeni pigmentozom retinitisa u početku imaju problema s vidom, posebno u slabo osvijetljenim okruženjima, i žale se na suženje perifernog vidnog polja. Središnji vid pošteđen je do kasnijih faza bolesti, a konačni ishod može dramatično varirati: mnogi ljudi s pigmentozom retinitisa zadržavaju ograničen vid tijekom svog života, dok drugi potpuno gube vid.
Retinitis pigmentosa je nasljedna bolest, uglavnom uzrokovana genetskim promjenama prenesenim od jednog ili oba roditelja. Vrsta genetskog defekta određuje koje su retinalne stanice najviše uključene u poremećaj i omogućuje razlikovanje, s kliničkog gledišta, različitih stanja. Do danas je identificirano više od 50 različitih genetskih defekata uključenih u pigmentozu retinitisa. Abnormalnosti se mogu prenijeti s roditelja na potomke putem jednog od tri nasljedna obrasca: autosomno recesivno, autosomno dominantno ili heterosomalno recesivno (vezano za X ili za X).
Simptomi
Za dodatne informacije: Retinitis Pigmentosa Simptomi
Retinitis pigmentosa obično se nalazi u adolescenata i mladih odraslih osoba. Simptomi se često pojavljuju u dobi od 10 do 30 godina, no dijagnoza se može postaviti u ranom djetinjstvu ili mnogo kasnije u životu.
Rani simptomi pigmentoze retinitisa mogu uključivati:
- Poteškoće s vidom noću (noćno sljepilo) ili u uvjetima slabog osvjetljenja
- Sporo prilagođavanje iz vida u mraku na ono u svjetlu i obrnuto;
- Suženje vidnog polja i gubitak perifernog vida;
- Osjetljivost na svjetlost i odsjaj.
Neki simptomi ovise o vrsti uključenih fotoreceptora. Štapovi su odgovorni za crno -bijeli vid, dok vam čunjevi omogućuju razlikovanje boja.
U većini slučajeva pigmentoznog retinitisa najprije se zahvaćaju štapići. Međutim, u oblicima koji se brzo razvijaju, na čunjeve se također može utjecati u ranoj fazi.
Štapići su koncentrirani u vanjskim dijelovima retine i aktiviraju se prigušenim svjetlom, pa njihova degeneracija utječe na periferni i noćni vid. Ako su u pitanju češeri, moguće je doživjeti gubitak percepcije boje i središnjeg vida.
Prevladavanje uključenih fotoreceptora određeno je posebnim nedostatkom prisutnim u genetskom sastavu pacijenta.
Često je prvi simptom pigmentoznog retinitisa noćno sljepilo (ili noktalopija). Neki ljudi smatraju da im treba sve više vremena da se prilagode razlikama u svjetlosti pri prelasku s dobro osvijetljenog područja na tamnije. Tipičan oblik gubitka vida izaziva sužavanje perifernog vida (vid u tunelu ili teleskopu); ovaj uzorak naziva se prstenasti skotom. Ponekad ovaj fenomen može nedostajati u ranim fazama, ali se primjećuje kada pojedinac često spotakne predmete ili je upleten u prometnu nesreću. Kada gubitak vida zahvaća središnje područje mrežnice (naziva se i makularna distrofija) doživjeti poteškoće s čitanjem i detaljnim radom koji zahtijeva koncentraciju na jedan predmet, kao što je provlačenje niti kroz ušice igle Mnogi pacijenti izvještavaju da su vidjeli bljeskove svjetlosti (fotopsiju), često opisane kao mala, trepereća i svjetlucava svjetla.
Brzina napredovanja bolesti i stupanj gubitka vida razlikuju se od osobe do osobe. Neki ekstremni slučajevi mogu se brzo razvijati u roku od dva desetljeća, drugi polako koji nikada ne dovodi do potpunog sljepila. Rani početak javlja se u težih oblika pigmentoznog retinitisa, dok bolesnici s blažim stanjima (npr. Autosomno dominantni) mogu razviti bolest u petom ili šestom desetljeću života. U obiteljima s X-vezanim pigmentoznim retinitisom muškarci su češće oboljeli nego žene i teže; žene, s druge strane, prenose genetske karakteristike (nose izmijenjeni gen na X kromosomu) i rjeđe manifestiraju simptome poremećaja.
Komplikacije
Retinitis pigmentosa će nastaviti napredovati, iako sporo. Međutim, potpuna sljepoća je rijetka, ali može doći do značajnog smanjenja perifernog i središnjeg vida.
Bolesnici s pigmentozom retinitisa često u ranoj dobi razviju oticanje mrežnice (makularni edem) ili kataraktu. Ove se komplikacije mogu liječiti ako ometaju vid.
Srodne bolesti
Uobičajeno, pacijent s pigmentozom retinitisa nema drugih poremećaja, au ovom slučaju govorimo o "nesindromskom" ili jednostavnom pigmentoznom retinitisu. Međutim, nekoliko sindroma dijeli neke kliničke simptome s ovom očnom bolešću; najčešći je Usherov sindrom, koji pogađa približno 10-30% svih pacijenata s pigmentozom retinitisa i povezan je s istodobnim urođenim ili progresivnim gubitkom sluha. U Leberovoj kongenitalnoj amaurozi, međutim, djeca mogu oslijepiti ili gotovo oslijepiti u prvih šest mjeseci života. Druge bolesti povezane s pigmentozom retinitisa uključuju Bardet-Biedlov sindrom i Refsum-ovu bolest.
Uzroci
Bolest može biti uzrokovana brojnim genetskim defektima: zapravo, postoji nekoliko gena koji, ako su zahvaćeni promjenom, mogu uzrokovati fenotip retinitis pigmentosa. Oni normalno kodiraju proteine uključene u kaskadu transdukcije koja omogućuje vid, utječe na transkripciju stanica (koje šalju pogrešne poruke stanicama retine) ili za elemente koji čine strukturu fotoreceptora. Naslijeđene genske mutacije prisutne su u stanicama od trenutka začeća; uobičajene abnormalnosti uključuju one gena RP1 (u retinitis pigmentosa-1, autosomno dominantni) , RHO (RP4, autosomno dominantno) i RDS (RP7, autosomno dominantno). Nenasljedni uzroci pigmentoze retinitisa su rijetki, ali postoji mogućnost pronalaska izoliranog slučaja (spontana mutacija), u kojem nije prisutna obiteljska anamneza bolest.




.jpg)

.jpg)

-cos-cause-e-disturbi-associati.jpg)