Što je fenilketonurija
Tamo fenilketonurija (P.K.U.) to je autosomno recesivna nasljedna metabolička bolest koja pogađa 1 na 10 000 osoba i čini se da se javlja više u homozigotnosti nego u heterozigota.
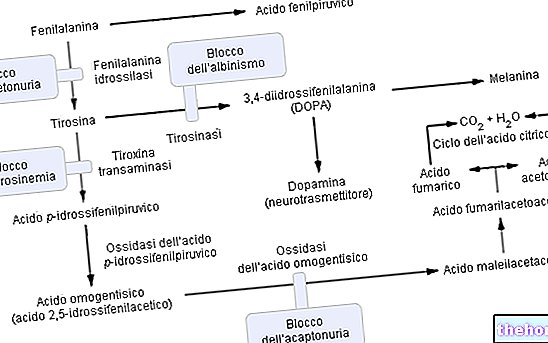
Pripada skupini hiperfenilalaninemije, fenilketonurija značajno kompromitira metabolizam fenilalanina, a posebno njegovu pretvaranje u tirozin; fenilketonurija se prepoznaje po povišenim razinama fenilalanina i nekih derivata u urinu (fenilpiruvat, fenilacetat, fenilaktat i fenilacetilglutamin).
Najozbiljnija komplikacija fenilketonurije je mentalno kašnjenje.
Fenilalanin, tirozin i derivati
Fenilalanin je esencijalna aminokiselina i čini većinu proteina u prehrani; može se pretvoriti enzimom fenilalanin hidroksilaza u tirozinu (dodavanjem hidroksilne skupine -OH). S druge strane, tirozin je aminokiselina prekursor za sintezu:
- L-DOPA (međuprodukt sinteze dopamina)
- Epinefrin
- Norepinefrin (svi neurotransmiteri).

Mehanizam fenilketonurije (P.K.U.)
Kao što se očekivalo, u fenilketonuriji, zbog jedne ili više (ukupno 6) kromosomskih mutacija, ekspresija (dakle i metabolička aktivnost) fenilalanin hidroksilaze je praktički nikakva. Te promjene mogu biti različitih vrsta (od "pogrešnih" promjena do "spajanja" defekata ili čak "djelomičnih brisanja"), ali ono što je važno je da zbog ove enzimske neučinkovitosti razina fenilalanina u krvi (koja je obično 1 mg / 100 ml) u DOMINANTNOJ fenilketonuriji lako dosežu količine čak 50 puta veće.
Djelovanje enzima fenilalanin hidroksilaze: Za proizvodnju tirozina (+ dihidrobiopterin), fenilalanin hidroksilazi su potrebni: fenilalanin, kisik i tetrahidrobiopterin (reducirani pteridin koji djeluje kao kofaktor); reakcija je također reverzibilna i dihidrobiopterin se može ponovno pretvoriti (zahvaljujući enzimu) dihidropterin reduktaza) u tetrahidrobiopterinu.
Komplikacije
Fenilketonurija može uzrokovati manje ili više ozbiljne komplikacije ovisno o ozbiljnosti patološke manifestacije i pravovremenosti postavljanja dijagnoze; kao nasljedna patologija, fenilketonurija se razlikuje po:
- Dominantna, stoga karakterizirana POTPUNOM neaktivnošću enzima fenilalanin hidroksilaze
- Recesivno, u kojem je aktivno samo 30% ukupnog enzimskog naslijeđa.
Komplikacije fenilketonurije mogu se pripisati i izravno proporcionalne metaboličkoj akumulaciji fenilalanina, njegovih derivata i smanjenoj sintezi tirozina. U patologiji se višak fenilalanina relativno učinkovito filtrira putem bubrega koji ga samo djelomično resorbira, uklanjajući ga urinom ; međutim, postojanost razina hiperfenilalaninemije određuje metaboličku reakciju molekularne PREVRTKE u fenilpiruvična kiselina i / ili druge derivate koji se lakše isušuju (fenilpiruvat, fenilacetat, fenilaktat).
Ono što komplicira fenilketonuriju je toksičnost fenilalanina, fenilpiruvične kiseline i njezinih derivata prema središnjem živčanom sustavu (CNS). Njihova prekomjerna prisutnost u razvoju mozga neumitno određuje oblik mentalne retardacije.
Napomena: Koncentracije ostalih aminokiselina u plazmi su blago smanjene, vjerojatno zbog povratne informacije o crijevnoj apsorpciji ili reapsorpciji bubrežnih tubula.
Oštećenje mozga, kao ozbiljna komplikacija fenilketonurije, uzrokovano je oduzimanjem drugih esencijalnih aminokiselina u proteosintezi, osobito u stvaranju poliribosoma, mijelina, noradrenalina i serotonina. Fenilketonurija - nije vidljiva odmah nakon rođenja, ali nakon nekoliko godina - ako se ne liječi, zahtijeva hospitalizaciju djeteta i potpuno je nepovratna.
Napredna fenilketonurija također može biti jasno vidljiva golim okom; visoke koncentracije fenilalanina, inhibirajući enzim tirozinaza, značajno umanjuju sintezu melanina smanjujući pigmentaciju kože i kose; nadalje, nakupljanje fenilacetata u kosi i koži daje fenilketonuricima snažan i neugodan "miris miša".




-cos-come-si-calcola-a-cosa-serve.jpg)


-contro-lobesit.jpg)